AACR Annual Meeting 2017: Challenging the Dogma of Treating IDH-mutant Cancers With IDH Inhibitors
Two studies presented at the AACR Annual Meeting 2017, one of which was also published in the AACR’s journal Cancer Research on April 1, showed that tumors that have mutations in the proteins isocitrate dehydrogenase-1 or -2 (IDH1/2) exhibited features similar to that of BRCA-mutant tumors and are, therefore, more likely to respond better to PARP inhibitors than to IDH inhibitors.
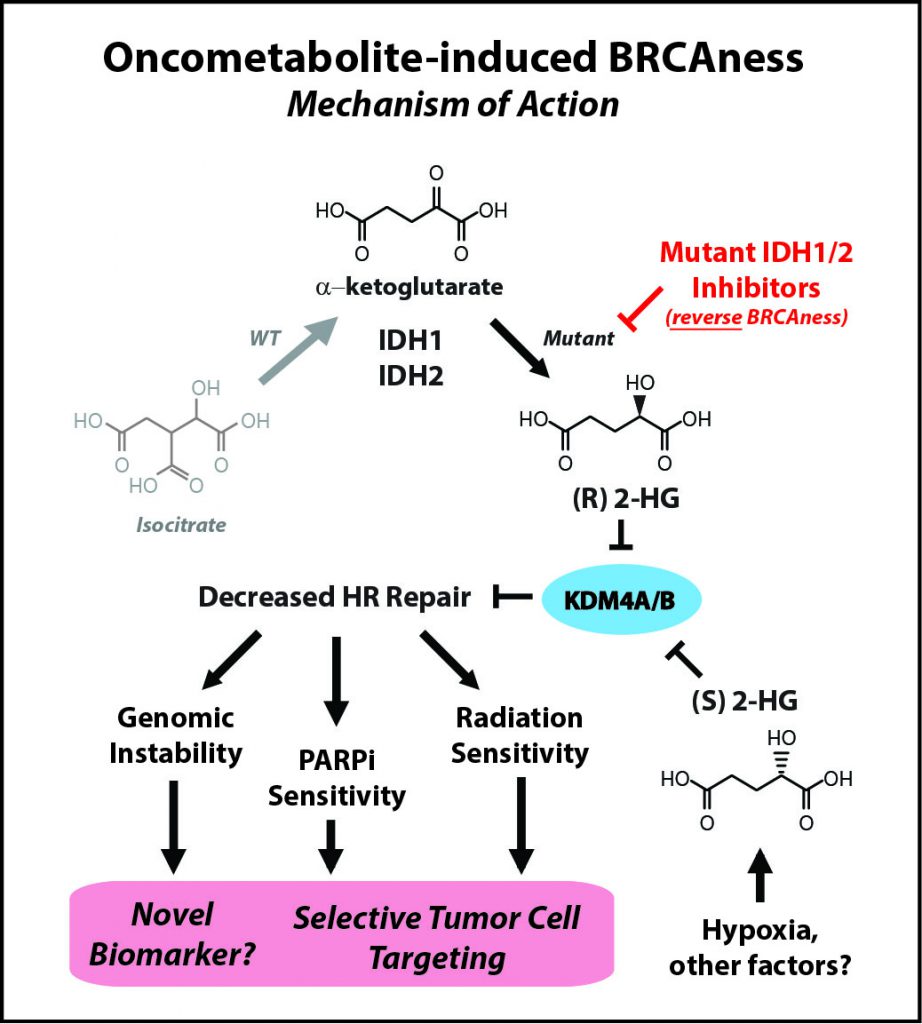
IDH mutations—commonly found in brain tumors, a devastating disease with an urgent need for better therapies—are neomorphic, meaning that they change the normal function of the encoded protein, explained Ranjit Bindra, MD, PhD, assistant professor of Therapeutic Radiology and Experimental Pathology at Yale School of Medicine and Yale Cancer Center in New Haven, Connecticut, who presented one of the studies at the Annual Meeting. Instead of making molecules that produce energy in the cell, which is the normal function of IDH, mutant IDH proteins start making an oncometabolite, 2-hydroxyglutarate (2HG), which is thought to be important for tumorigenesis.

Ranjit Bindra, MD, PhD
Based on this knowledge, therapeutics that block the mutant IDH1 and IDH2 proteins and decrease the levels of 2HG are being developed and tested in clinical trials. However, it is not entirely clear whether these IDH inhibitors have been effective in shrinking solid tumors, according to Bindra.
“The remarkable thing is that we are proposing a therapy which does exactly the opposite of what IDH inhibitors are designed to do,” Bindra said.
“We made an unexpected discovery that 2HG, both made by hypoxia and by IDH1 [the two enantiomers of 2HG, namely R-2HG and S-2HG], are potent suppressors of homologous [DNA] recombination,” Bindra said. “This essentially makes them quite similar to breast and ovarian cancers that harbor mutations in key DNA repair genes such as BRCA1 and BRCA2.”
IDH inhibitors reverse BRCAness
Laboratory research led by Bindra’s team showed that by suppressing DNA repair, the IDH mutations essentially create an Achilles’ heel in the tumor cell, and that this weakness can be exploited by treating these tumors with PARP inhibitors, instead of IDH inhibitors. “In fact, small molecule inhibitors of mutant IDH actually reverse PARP inhibitor sensitivity,” said Bindra.
A PARP inhibitor is a DNA repair inhibitor, and when applied to a tumor that has a DNA repair defect, the effects of the defect and inhibitor are synergistic, as a result of a process known as “synthetic lethality,” leading to tumor cell death. Therapeutics developed based on this approach have been effective in treating hereditary breast and ovarian cancers, and the U.S. Food and Drug Administration has approved two PARP inhibitors, olaparib (Lynparza) and rucaparib (Rubraca), to treat certain BRCA-mutant ovarian cancers, and a third PARP inhibitor, niraparib (Zejula), to treat recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancers regardless of BRCA status.
Bindra’s team showed that IDH-mutant patient-derived glioma cells, bone-marrow cultures of IDH-mutant acute myeloid leukemia, and mice bearing IDH-mutant human tumor cells were all susceptible to PARP inhibition. Results from these studies were published recently in Science Translational Medicine.
The investigators conducted further experiments in genetically matched IDH-wild type (normal form of the protein) and IDH-mutant cancer cell line models to compare the efficacy of IDH inhibitors and PARP inhibitors in inhibiting IDH-mutant tumor cell viability. “When you carefully control the conditions and simply ask the question of which drugs best target solid tumor cell lines with IDH mutations, we do not see a significant effect of mutant IDH inhibitors on viability, however, we do see profound differences with PARP inhibitors,” said Bindra.
“This has revolutionary implications,” said Louis Weiner, MD, who moderated the press conference. The reflex approach of oncologists is to treat a tumor driven by IDH mutations with an IDH inhibitor; however, data presented by Bindra suggest that “it may be exactly the wrong thing to do,” Weiner said. “The correct way is not to think that all mutations are drivers that should be inhibited, but rather that there are mutations that are actually creating vulnerabilities that can be exploited.”
Several other studies support the hypothesis
“We are not the only laboratories that see this phenotype,” Bindra said during his presentation, citing data from a study published in the AACR’s journal Molecular Cancer Research, in which the authors found that IDH1 mutations, which drive the development of glioma, rapidly convert from being the driver of the disease to a passenger. “Although mutant IDH1 expression is thought to drive the gliomagenesis process, the extent to which it remains a viable therapeutic target remains unknown,” the authors of the paper wrote.
In the other presentation at the AACR Annual Meeting 2017, a team of researchers from the National Cancer Institute (NCI) and the University of North Carolina found that IDH1-mutated glioma cells had increased DNA damage, and underproduced an important substrate for PARP-associated DNA repair, which may make them more sensitive to PARP inhibitors. In their Cancer Research paper, they also showed that targeting the PARP-associated DNA repair pathway sensitized the IDH-mutant glioma cells to temozolamide, a chemotherapeutic commonly used to treat this disease.
Bindra’s team received a conditional approval by the NCI to initiate a multicenter, phase II trial across approximately 35 centers in the United States, in which they will test olaparib in patients with IDH-mutant cancers.